スキャン動画で画像解析による形質評価
良くあるハイスループットなフェノタイピングとしてベルトコンベアを用いたものや専用のレールに取り付けられたカメラを用いたもの、ドローンを用いたものなどがあると思います。ただ、どれも多少の導入コストがかかるため、1回きりの調査には使いづらい感があります。
特に個人ではなかなか手が出しづらいです。
そこで、スマホとその辺にある台車をつかって手軽にデータを取れないか試してみました。
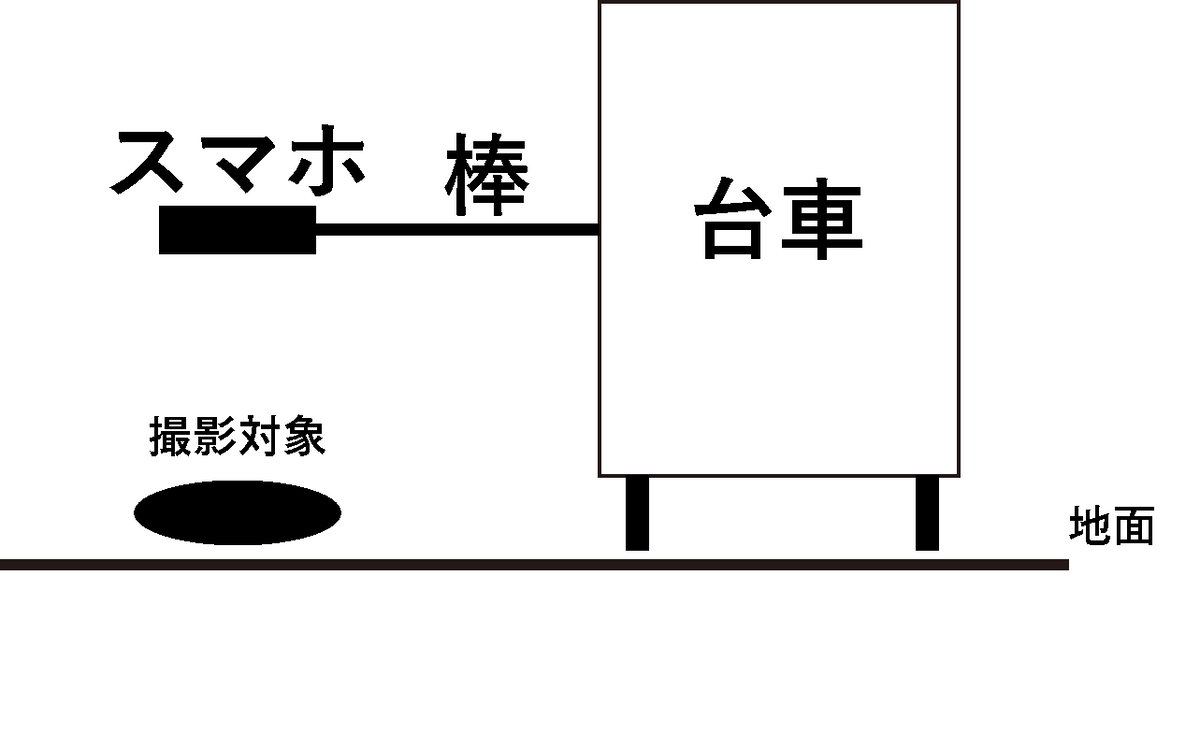
具体的には、以下の図に示すような装置を作って、装置を手で速度を一定に保ちながら動かして、対象の動画を撮影しました。
今回は対象として徳島県産と長崎県産のニンジンを使いました。それぞれのニンジンの産地がわかるように各サンプルの左端には1辺10cmで産地名をコードしたQRを配置しました。
イメージはこのような感じです。
実際に撮影された動画はこのようになりました。

動画の各フレームからQRとニンジンを検出する
動画は複数の連続する画像(フレーム)からなっているので各フレームからQRとニンジンを検出していきます。opencvを用いて動画から各フレームを読み込んで検出、結果をディスプレイに表示、ファイルに書き込みという一連の処理は、以下のループで実装しました。
video_path = "input.mp4" cap = cv2.VideoCapture(video_path) if not cap.isOpened(): raise IOError(f"Cannot open video file: {video_path}") # 動画の各種情報を取得する frame_count = cap.get(cv2.CAP_PROP_FRAME_COUNT) frame_width = cap.get(cv2.CAP_PROP_FRAME_WIDTH) frame_height = cap.get(cv2.CAP_PROP_FRAME_HEIGHT) fps = cap.get(cv2.CAP_PROP_FPS) # 検出結果の保存するための書き込み用オブジェクトも作っておく fourcc = cv2.VideoWriter_fourcc(*'mp4v') out = cv2.VideoWriter('output.mp4', fourcc, fps, (int(frame_width), int(frame_height))) # 動画のフレームをループして1つずつ処理する while cap.isOpened(): # 動画から1フレーム読み込む success, frame = cap.read() pos_frames = cap.get(cv2.CAP_PROP_POS_FRAMES) if success: # frameに対してメイン処理 # 検出結果を描画したフレームを出力ファイルに書き込む out.write(frame) # 同じ情報をディスプレイ上にも表示する cv2.imshow("ニンジントラッキング", frame) # qキーが押下されたら終了する if cv2.waitKey(1) & 0xFF == ord("q"): break
QRの検出
QRについての検出はopencvのQRCodeDetectorArucoを用いました。
qr_detector = cv2.QRCodeDetectorAruco() qr_detected, decoded_info_list, corners_list, _ = qr_detector.detectAndDecodeMulti(frame)
ニンジンの検出
ニンジンに検出にはyolo26を用いました。Ultralyticsによって開発されているライブラリに含まれています。かなりしっかりとメンテナンスされているいんしょうで、ホビー用途では無料で使えるのでありがたいです。
ニンジンの検出は少し特殊で、1フレームに複数映る可能性があるので、各個体を識別しながら検出する必要があります。 そこで今回は各検出のトラッキングを行い、同一個体と予測されたものに識別可能なIDを振る処理を行いました。
UltralyticsのYOLOを用いる場合はすごく簡単でmodel.track()でそれが可能です。
model = YOLO("yolo26n-seg.pt") result = model.track(frame, conf=0.8, iou=0.8, persist=True)[0]
検出についてをまとめたコード全体
以下に全体的なコードを示します。各フレームで検出されたQRコードとニンジンの情報を格納し、最後にデータフレームにしています。また、検出状況はディスプレイに表示されるようにしていますが、後で見返しもできるように動画に記録もしています。
コードは以下を参考にしました。
今回はニンジンの根長と根径を検出時のバウンディングボックスの高さと幅で大まかに近似して測ることにしています。
from ultralytics import YOLO from collections import defaultdict import cv2 import numpy as np from ultralytics import YOLO from dataclasses import dataclass import pandas as pd @dataclass class Carrot: id: int x: float y: float w: float h: float pos_frames: float @dataclass class QRCode: decoded_info: str x: float y: float side_length: float pos_frames: float # YOLO26モデルをロードする(今回はニンジンをこのモデルを使って検出する) model = YOLO("yolo26n-seg.pt") # OpenCVのQR検出器を作成する qr_detector = cv2.QRCodeDetectorAruco() # 動画を開く video_path = "input.mp4" cap = cv2.VideoCapture(video_path) if not cap.isOpened(): raise IOError(f"Cannot open video file: {video_path}") # 動画の各種情報を取得する frame_count = cap.get(cv2.CAP_PROP_FRAME_COUNT) frame_width = cap.get(cv2.CAP_PROP_FRAME_WIDTH) frame_height = cap.get(cv2.CAP_PROP_FRAME_HEIGHT) fps = cap.get(cv2.CAP_PROP_FPS) print(f"総フレーム数: {frame_count}, フレーム幅: {frame_width}, フレーム高さ: {frame_height}, FPS: {fps}") # 検出結果の保存するための書き込み用オブジェクトも作っておく fourcc = cv2.VideoWriter_fourcc(*'mp4v') out = cv2.VideoWriter('output.mp4', fourcc, fps, (int(frame_width), int(frame_height))) # 検出されたQRコードとニンジンの保存のためのリスト qr_codes = [] carrots = [] # 動画のフレームをループして1つずつ処理する while cap.isOpened(): # 動画から1フレーム読み込む success, frame = cap.read() pos_frames = cap.get(cv2.CAP_PROP_POS_FRAMES) if success: # YOLOを実行して対象をトラッキングする result = model.track(frame, conf=0.8, iou=0.8, persist=True)[0] # 得られたバウンディングボックスがあれば処理を開始 if result.boxes and result.boxes.is_track: boxes = result.boxes.xywh.cpu() track_ids = result.boxes.id.int().cpu().tolist() classes = result.boxes.cls.int().cpu().tolist() # 対象検出結果をフレームに描画する frame = result.plot() # 検出された物体を1つずつ処理する for box, track_id, class_id, in zip(boxes, track_ids, classes): if model.names[class_id] != 'carrot': # ニンジン以外はスキップ continue x, y, w, h = box # ニンジン検出結果を保存 carrots.append(Carrot( id=track_id, x=float(x), y=float(y), w=float(w), h=float(h), pos_frames=pos_frames )) # ニンジンの中心座標にマーカーを描画する cv2.drawMarker(frame, (int(x), int(y)), (255, 255, 0), 1, 50, 10) # QRコード検出を行う qr_detected, decoded_info_list, corners_list, _ = qr_detector.detectAndDecodeMulti(frame) if qr_detected: for decoded_info, corners in zip(decoded_info_list, corners_list): if decoded_info == "": # デコードされた結果が空文字列の場合はスキップ continue # QRの中心座標を計算して描画する(コーナー座標の平均) x, y = corners.mean(axis=0) cv2.drawMarker(frame, (int(x), int(y)), (255, 255, 0), 1, 50, 10) # QRの辺の長さを計算する side_length = np.mean(np.linalg.norm(corners - np.roll(corners, shift=1, axis=0), axis=1)) # QR検出結果を保存 qr_codes.append(QRCode( decoded_info=decoded_info, x=x, y=y, side_length=side_length, pos_frames=pos_frames )) # QRの検出結果を描画する corners = corners.astype(int) for i in range(len(corners)): cv2.line(frame, tuple(corners[i]), tuple(corners[(i + 1) % len(corners)]), (0, 255, 0), 10) if decoded_info: cv2.putText(frame, decoded_info, (corners[0, 0], corners[0, 1] - 10), cv2.FONT_HERSHEY_SIMPLEX, 3, (0, 255, 0), 10) # 検出結果を描画したフレームを出力ファイルに書き込む out.write(frame) # 同じ情報をディスプレイ上にも表示する cv2.imshow("ニンジントラッキング", frame) # qキーが押下されたら終了する if cv2.waitKey(1) & 0xFF == ord("q"): break else: # 動画が最後に到達したら終了する break # 動画キャプチャオブジェクトを解放して結果表示ウィンドウを閉じる cap.release() out.release() cv2.destroyAllWindows() # 結果をデータフレームに保存する df_carrots = pd.DataFrame(carrots) df_qr_codes = pd.DataFrame(qr_codes)
総フレーム数: 600.0, フレーム幅: 2160.0, フレーム高さ: 3840.0, FPS: 60.0
検出の様子
結果は以下のGIF動画に示すようになりました。

検出したデータから目的の形質を集計する
ニンジン検出結果は以下のようになり1000フレーム以上から結果が得られていることになました。YOLOのトラッキング機能によって各検出にはIDが振られており、同一IDイコール同一個体として識別可能になります。また、各検出の中心座標(x, y)、バウンディングボックスの幅と高さ(w, h)と検出されたフレーム位置(何番目のフレームか)(pos_frames)を今回は記録しています。
df_carrots
| id | x | y | w | h | pos_frames | |
|---|---|---|---|---|---|---|
| 0 | 1 | 1949.486328 | 1463.835205 | 421.027344 | 1487.324707 | 1.0 |
| 1 | 1 | 1949.034424 | 1463.601929 | 421.827026 | 1487.832764 | 2.0 |
| 2 | 1 | 1948.967529 | 1463.503540 | 421.823364 | 1488.549561 | 3.0 |
| 3 | 1 | 1948.907471 | 1462.683838 | 421.866211 | 1488.790039 | 4.0 |
| 4 | 1 | 1948.970947 | 1461.983032 | 421.774292 | 1488.717041 | 5.0 |
| ... | ... | ... | ... | ... | ... | ... |
| 1001 | 5 | 687.110168 | 2273.889160 | 365.511963 | 2044.912231 | 599.0 |
| 1002 | 6 | 1145.700928 | 1989.584961 | 387.035339 | 1371.399048 | 599.0 |
| 1003 | 4 | 199.110947 | 2048.687012 | 379.045929 | 1717.308960 | 600.0 |
| 1004 | 5 | 677.186340 | 2278.349609 | 364.560333 | 2052.345947 | 600.0 |
| 1005 | 6 | 1134.652466 | 1989.436890 | 385.959473 | 1371.355713 | 600.0 |
1006 rows × 6 columns
QRコードについても同様に見てみると160フレーム以上から検出ができていて、同様に中心座標(x, y)やフレーム位置(pos_frames)を記録しています。バウンディングボックス情報は記録していないのとID情報はありませんが、その代わりに、デコードされた情報、辺の長さ(px単位)を記録しています。
df_qr_codes
| decoded_info | x | y | side_length | pos_frames | |
|---|---|---|---|---|---|
| 0 | tokushima | 581.683594 | 1547.049805 | 859.927246 | 1.0 |
| 1 | tokushima | 581.452271 | 1547.780884 | 861.898560 | 2.0 |
| 2 | tokushima | 580.443970 | 1547.052734 | 860.664185 | 3.0 |
| 3 | tokushima | 580.452026 | 1547.276855 | 860.923462 | 4.0 |
| 4 | tokushima | 579.443665 | 1546.787354 | 860.894775 | 5.0 |
| ... | ... | ... | ... | ... | ... |
| 160 | nagasaki | 500.842163 | 1742.364380 | 862.768433 | 446.0 |
| 161 | nagasaki | 489.627319 | 1743.115723 | 862.798096 | 447.0 |
| 162 | nagasaki | 480.617737 | 1742.910400 | 865.022095 | 448.0 |
| 163 | nagasaki | 472.753601 | 1745.186157 | 864.993408 | 449.0 |
| 164 | nagasaki | 465.642639 | 1745.708862 | 864.330017 | 450.0 |
165 rows × 5 columns
各ニンジン個体がどのフレームで検出されたかをグラフにすると以下のようになります。
import matplotlib.pyplot as plt fig, ax = plt.subplots() ax.plot(df_carrots.pos_frames, [f"carrot:{i}" for i in df_carrots.id], 'C1o') plt.show()
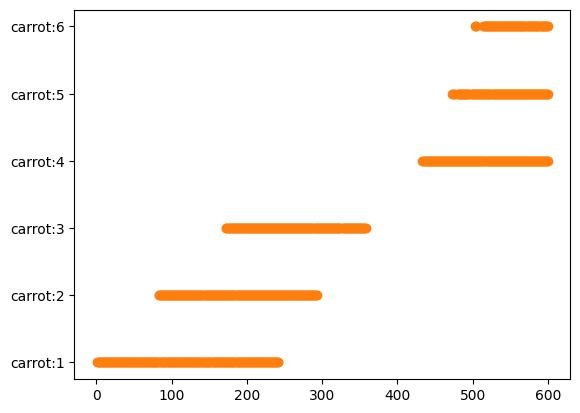
ここで、フレームの端っこで検出されたニンジンは端っこが切れていたり、切れていなくても歪んでいたりする可能性があるので、それらを除きたいと思います。今回はニンジンの中心のx座標が動画フレームの中心10%に入るデータだけを切り出して解析に用いたいと思います。
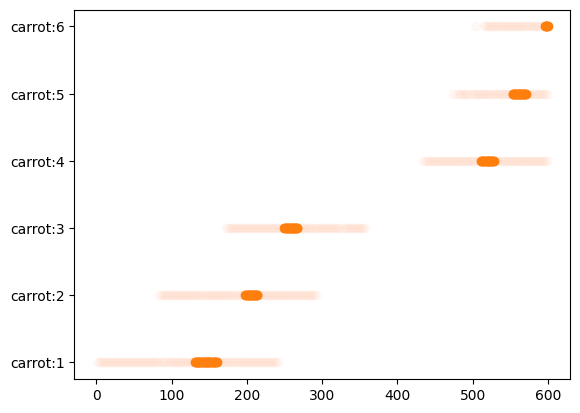
中央10%に入るニンジンフレームからの情報だけ切り出す
ここで濃いオレンジでプロットした部分が残ったデータになります。
index = np.logical_and(df_carrots.x > frame_width * 0.45, df_carrots.x < frame_width * 0.55) df_carrots_center = df_carrots[index] fig, ax = plt.subplots() ax.plot(df_carrots.pos_frames, [f"carrot:{i}" for i in df_carrots.id], 'C1o', alpha=.01) ax.plot(df_carrots_center.pos_frames, [f"carrot:{i}" for i in df_carrots_center.id], 'C1o') plt.show()
各ニンジンの平均情報を算出する
動画フレームごとに検出などにノイズが入ることから、検出高さと幅などの平均を取ることとします。また、この時点で検出された平均フレームも取得します。
df_carrots_mean = df_carrots.groupby('id').mean().reset_index()
df_carrots_mean
| id | x | y | w | h | pos_frames | |
|---|---|---|---|---|---|---|
| 0 | 1 | 1206.867914 | 1494.401272 | 498.538615 | 1475.519982 | 121.299578 |
| 1 | 2 | 1182.876424 | 1599.698061 | 450.049639 | 1567.755488 | 188.234450 |
| 2 | 3 | 1051.950533 | 1568.823529 | 472.428185 | 1562.927545 | 264.211957 |
| 3 | 4 | 1144.410894 | 2046.994780 | 384.317635 | 1678.992444 | 516.987952 |
| 4 | 5 | 1352.268076 | 2288.102073 | 348.824204 | 2046.767100 | 538.696721 |
| 5 | 6 | 1615.287771 | 1993.774678 | 387.900986 | 1374.018268 | 554.670455 |
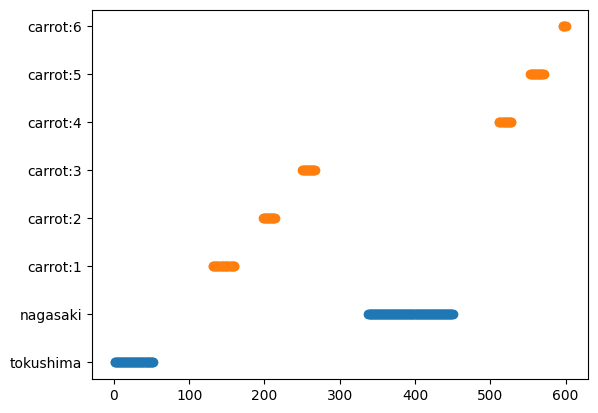
QRコードが出現したフレームと並べる
QRコードが検出されたフレームの情報を並べて表示してみると順番に検出できていることがわかります。各QRコードの平均フレームを算出し、それ以降に出現したニンジンにそのQRでコードされている情報(ここでは産地ですが、系統名などでも良いでしょう)を割り振りつつ、個体IDも産地ごとの動画での出現順に振りなおしたいと思います。
index = np.logical_and(df_qr_codes.x > frame_width * 0.2, df_qr_codes.x < frame_width * 0.8) fig, ax = plt.subplots() ax.plot(df_qr_codes[index].pos_frames, df_qr_codes[index].decoded_info, 'C0o') ax.plot(df_carrots_center.pos_frames, [f"carrot:{i}" for i in df_carrots_center.id], 'C1o') plt.show()
df_qr_codes_mean = df_qr_codes.groupby('decoded_info').mean().reset_index() df = pd.concat([df_carrots_mean, df_qr_codes_mean]) df.sort_values('pos_frames', inplace=True) df['type'] = np.where(df.decoded_info.isna(), 'carrot', 'QR') df[['decoded_info', 'side_length']] = df[['decoded_info', 'side_length']].ffill() df['individual_id'] = df.groupby('decoded_info').cumcount() df = df.reset_index(drop=True) df
| id | x | y | w | h | pos_frames | decoded_info | side_length | type | individual_id | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | NaN | 550.764160 | 1546.536377 | NaN | NaN | 26.500000 | tokushima | 861.869568 | QR | 0 |
| 1 | 1.0 | 1206.867914 | 1494.401272 | 498.538615 | 1475.519982 | 121.299578 | tokushima | 861.869568 | carrot | 1 |
| 2 | 2.0 | 1182.876424 | 1599.698061 | 450.049639 | 1567.755488 | 188.234450 | tokushima | 861.869568 | carrot | 2 |
| 3 | 3.0 | 1051.950533 | 1568.823529 | 472.428185 | 1562.927545 | 264.211957 | tokushima | 861.869568 | carrot | 3 |
| 4 | NaN | 1079.954102 | 1737.297241 | NaN | NaN | 394.000000 | nagasaki | 851.076233 | QR | 0 |
| 5 | 4.0 | 1144.410894 | 2046.994780 | 384.317635 | 1678.992444 | 516.987952 | nagasaki | 851.076233 | carrot | 1 |
| 6 | 5.0 | 1352.268076 | 2288.102073 | 348.824204 | 2046.767100 | 538.696721 | nagasaki | 851.076233 | carrot | 2 |
| 7 | 6.0 | 1615.287771 | 1993.774678 | 387.900986 | 1374.018268 | 554.670455 | nagasaki | 851.076233 | carrot | 3 |
検出されたQRコードの辺の長さをリファレンスとして根長と根径をcm単位で測定
今回用いたQRコードは1辺が10cmであることがわかっているのでその情報を元に、検出されたニンジンの根長と根径をそれぞれバウンディングボックスの高さと幅で近似して測定します。
df['diameter_cm']= df['w'] / df['side_length'] * 10 df['length_cm']= df['h'] / df['side_length'] * 10 df
| id | x | y | w | h | pos_frames | decoded_info | side_length | type | individual_id | diameter_cm | length_cm | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | NaN | 550.764160 | 1546.536377 | NaN | NaN | 26.500000 | tokushima | 861.869568 | QR | 0 | NaN | NaN |
| 1 | 1.0 | 1206.867914 | 1494.401272 | 498.538615 | 1475.519982 | 121.299578 | tokushima | 861.869568 | carrot | 1 | 5.784386 | 17.119992 |
| 2 | 2.0 | 1182.876424 | 1599.698061 | 450.049639 | 1567.755488 | 188.234450 | tokushima | 861.869568 | carrot | 2 | 5.221784 | 18.190171 |
| 3 | 3.0 | 1051.950533 | 1568.823529 | 472.428185 | 1562.927545 | 264.211957 | tokushima | 861.869568 | carrot | 3 | 5.481435 | 18.134154 |
| 4 | NaN | 1079.954102 | 1737.297241 | NaN | NaN | 394.000000 | nagasaki | 851.076233 | QR | 0 | NaN | NaN |
| 5 | 4.0 | 1144.410894 | 2046.994780 | 384.317635 | 1678.992444 | 516.987952 | nagasaki | 851.076233 | carrot | 1 | 4.515666 | 19.727874 |
| 6 | 5.0 | 1352.268076 | 2288.102073 | 348.824204 | 2046.767100 | 538.696721 | nagasaki | 851.076233 | carrot | 2 | 4.098625 | 24.049163 |
| 7 | 6.0 | 1615.287771 | 1993.774678 | 387.900986 | 1374.018268 | 554.670455 | nagasaki | 851.076233 | carrot | 3 | 4.557770 | 16.144479 |
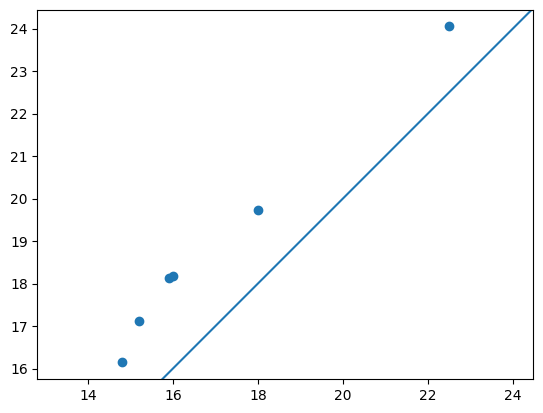
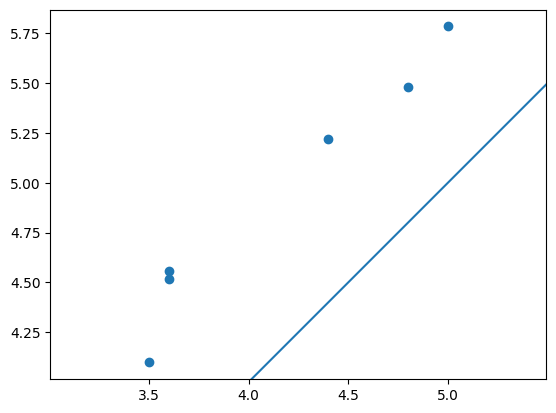
手計測の値と比較する
最後に、手計測で取得された根長と根径を今回の画像解析での測定値で比較してみます。
df_manual = pd.DataFrame({
'production_area': ['tokushima'] * 3 + ['nagasaki'] * 3,
'individual_id': [1, 2, 3] * 2,
'length_by_manual': [15.2, 16.0, 15.9, 18.0, 22.5, 14.8],
'diameter_by_manual': [5.0, 4.4, 4.8, 3.6, 3.5, 3.6],
})
df_manual
| production_area | individual_id | length_by_manual | diameter_by_manual | |
|---|---|---|---|---|
| 0 | tokushima | 1 | 15.2 | 5.0 |
| 1 | tokushima | 2 | 16.0 | 4.4 |
| 2 | tokushima | 3 | 15.9 | 4.8 |
| 3 | nagasaki | 1 | 18.0 | 3.6 |
| 4 | nagasaki | 2 | 22.5 | 3.5 |
| 5 | nagasaki | 3 | 14.8 | 3.6 |
df = df.rename(columns={'decoded_info': 'production_area'})
df = df.set_index(['production_area', 'individual_id']).join(df_manual.set_index(['production_area', 'individual_id']))
plt.scatter(
x=df['length_by_manual'],
y=df['length_cm']
)
plt.axline(xy1=[df['length_by_manual'].mean()]*2, slope=1)
plt.axis('equal')
plt.show()
plt.scatter(
x=df['diameter_by_manual'],
y=df['diameter_cm']
)
plt.axline(xy1=[df['diameter_by_manual'].mean()]*2, slope=1)
plt.axis('equal')
plt.show()
df = df['length_cm'] - df['length_by_manual'])
production_area individual_id
tokushima 0 NaN
1 1.919992
2 2.190171
3 2.234154
nagasaki 0 NaN
1 1.727874
2 1.549163
3 1.344479
dtype: float64
df['diameter_cm'] - df['diameter_by_manual']
production_area individual_id
tokushima 0 NaN
1 0.784386
2 0.821784
3 0.681435
nagasaki 0 NaN
1 0.915666
2 0.598625
3 0.957770
dtype: float64
まとめ
- スマホを移動する形式で撮影した画像からQRコード検出、計測対象物体検出は可能
- QRをサイズリファレンスとして手計測値と相関するデータは得られそう
- ただし今回の設定・手法だと1~2cmほど大きく見積もってしまう
今回生じた誤差に関しては以前書いた記事にある内容も関係しているかもしれません。
参考
Grid Engine系ジョブスケジューラー上でsnakemakeを走らせる
snakemakeコマンドを実行するラッパースクリプトをqsubでGrid Engineに投げるのではなく、executorとして(Sun/Univa/Altar) Grid Engineを指定する方法を紹介します。
私の環境ではUniva Grid Engineが動いているのでそちらでテストしていますが、多くの場合はそのほかの互換性のあるGrid Engineでも動かせるはずです。以後、Grid Engine系ジョブスケジューラーのことをSGEと呼びます。
snakemakeはバージョンが変わるごとにexecutorの仕様が結構変わっており、追いかけるのが大変だったので、2026年1月時点のsnakemake 9での情報を書き留めておきます。
すべてのexecutor backendはプラグインに
昔のsnakemakeではexecutorを指定する機能もデフォルトでついてきていた記憶がありますが、現在はプラグインとして提供されています。
プラグインのドキュメントはこちらにあります。ただ、このドキュメントはあまり親切ではなく、設定が難しく感じました。いろいろ試行錯誤したので、備忘録として記します。
executor pluginのインストール
SGEそのものを対象としたプラグインは存在しないので、cluster-genericのプラグインをインストールします。
pip install snakemake-executor-plugin-cluster-generic
または
conda install -n snakemake snakemake-executor-plugin-cluster-generic
executorを指定してsnakemakeを実行
先ほどプラグインをインストールしたので、cluster-genericがexecutorオプションに指定できるようになっています。
早速、以下のように指定して実行してみます。
snakemake --executor cluster-generic
すると
The following required arguments are missing for plugin cluster-generic: --cluster-generic-submit-cmd.
と出るので、サブミットコマンド、つまりqsubを指定してもう一度実行してみます。
snakemake --executor cluster-generic --cluster-generic-submit-cmd qsub
すると、またエラーで、
Error: maximum number of parallel jobs/used nodes has to be specified for remote execution (use --jobs N with N being a number >= 1)
と言われるので、並列に実行する最大ジョブ数を適当に10と指定して、
snakemake --executor cluster-generic --cluster-generic-submit-cmd qsub --jobs 10
ここまでしてやっと動きました。
external jobidの整合性をとって、qdel可能にする
しかし、よくログを見てみると以下のような行が現れました。
Submitted job 6 with external jobid 'Your job 37472 ("snakejob.test_rule.6.sh") has been submitted'.
external jobidがYour job 37472 ("snakejob.test_rule.6.sh") has been submittedとなっているとのことです。external jobidとはSGE側で割り振られるjobidのことで、ここは整数であって欲しいところです。
ドキュメントではこの辺りの説明が乏しかったのですが、実装を見てみると、--cluster-generic-submit-cmdで指定したコマンドを実行したときに、標準出力に出力される文字列をそのままexternal jobidとして認識しているようでした。
私の環境ではオプションなしのqsubコマンドの出力は人が読みやすいメッセージ形式となっています。そのため、external jobidが文字列として認識されてしまっていると考えられます。
とはいえ、external jobidが文字列になってしまっている点以外は実行上問題ありませんでした。しかし、jobidが正確に認識されていないことを考えると、snakemake側でジョブのキャンセル等はうまくいかない気がしてきます。
また、--executorにcluster-genericを指定した際は、先ほどの--cluster-generic-submit-cmdのほかに、--cluster-generic-status-cmd、--cluster-generic-cancel-cmd、--cluster-generic-cancel-nargs、--cluster-generic-sidecar-cmdというオプションが必須ではないものの指定可能です。
そこで、試しに--cluster-generic-cancel-cmdオプションについても指定してみます。
snakemake --executor cluster-generic --cluster-generic-submit-cmd qsub --jobs 10 --cluster-generic-cancel-cmd qdel
このワークフローが実行中にCtrl+Cなどで実行を中断してみるとどうなるでしょうか?
すると、以下のようなログが出ました。
The job Your job 37493 ("snakejob.test_sleep.8.sh") has been submitted of user(s) test_user does not exist
1 out of 1 calls to --cluster-cancel failed. This is safe to ignore in most cases.
つまり、SGEにサブミットされたジョブのキャンセルが失敗したことがわかります。ジョブの内容が有限で終了するものであるのならば放っておいても構わないかもしれませんが、今回はこの問題の解決も試みてみます。
原因はおそらく、先ほどのexternal jobidの不具合と同様に、snakemakeがジョブスケジューラー側のjobidを適切に認識していないことにあります。
そこでqsubコマンドの標準出力がjobidのみというフォーマットになるようにしたいので、ちょうどよいqsubオプションを使います。それが-terseオプションで、以下のように再度snakemakeを実行してみます。
snakemake --executor cluster-generic --cluster-generic-submit-cmd 'qsub -terse' --jobs 10 --cluster-generic-cancel-cmd qdel
すると先ほど違和感のあった部分のログは以下のようになり、
Submitted job 7 with external jobid '37499'.
Ctrl+Cで終了した場合のログは以下のようになりました。
Terminating processes on user request, this might take some time. test_user has registered the job 37499 for deletion
(test_userと37499の部分は環境によって異なります)
これで、正常にジョブをキャンセルできているように見えます。
Snakefileで指定したthreadsの値をSGE予約スロット数に反映する
ここまででもだいぶ使えるようになったものの、この設定だけでは、ruleの定義などで設定したthreadsの値がqsub実行時の予約スロット数に反映できていません。
SGE系で並列ジョブを投げる場合にスロット数を指定するには並列環境と並列数をオプションで指定すればよいのですが、Snakefile内のthreadsはruleごとに異なります。そこで{}を使ったプレースホルダを指定すると動的に反映できます。
具体的には
snakemake --executor cluster-generic --cluster-generic-submit-cmd 'qsub -terse -pe threads {threads}' --jobs 10 --cluster-generic-cancel-cmd qdel
こうしておけば、ジョブスケジューラー側にも使用するスロット数を指定してジョブをサブミットできるようになり、意図せずクラスターを過負荷にすることを避けられるはずです。
そのほか、いろいろ現段階で指定しておくとよさそうなものを見ていきます。
ジョブの名前
qsubの-Nオプションにプレースホルダ付きで指定できます。
snakemake --executor cluster-generic --cluster-generic-submit-cmd 'qsub -terse -pe threads {threads} -N {rulename}.{jobid}' --jobs 10 --cluster-generic-cancel-cmd qdel
ログの出力先の指定
個人的にはログファイルでカレントディレクトリがいっぱいになるのが好きではないので、すべてlogs/sge配下に格納されるようにしておきます。
snakemake --executor cluster-generic --cluster-generic-submit-cmd 'qsub -terse -pe threads {threads} -N {rulename}.{jobid} -e logs/sge/' --jobs 10 --cluster-generic-cancel-cmd qdel
これまでの設定をprofileにまとめる
以下の記載を$HOME/.config/snakemake/sge/config.yamlとして保存することで、--profile sgeとして起動できます。並列ジョブ数だけはYAMLファイルから外して実行ごとに変更しやすいようにしておきます。
executor: cluster-generic cluster-generic-submit-cmd: 'qsub -terse -pe threads {threads} -N {rulename}.{jobid} -e logs/sge/' cluster-generic-cancel-cmd: qdel
実行の際は以下のようにしてファイルに記載した設定を読み出して実行できます。
snakemake --profile sge --jobs 10
参考
snakemakeでEnvironment Modulesをつかう
バイオインフォマティクスの解析を実行する際、ソフトウェアやパッケージのバージョンなどのコンフリクトを避けたり、簡単に切り替えたりするためにEnvironment Modulesを利用して環境を動的に変更することがあるかと思います。
また、バイオインフォマティクスの解析パイプラインをsnakemakeで定義して実行することもあるかと思います。
snakemakeからEnvironment Modulesで管理されているソフトウェアをロードするには、単にshell:の部分にmodule load <module name>を追加すればよいですが、もう少し洗練された方法にenvmodulesディレクティブを使う方法があります。
具体的には、
rule rule_with_envmodules:
envmodules:
"some_module_01/0.0.0",
"some_module_02/1.1.1",
といった感じで指定してから、snakemake --use-envmodulesというオプション付きで実行すると、当該ruleが実行される前に、指定されたモジュールが上から順にmodule loadされるようです。
利用方法はかなり簡便ですが、使っているうちにいくつか注意点があるなと思ったので備忘録的に記しておきます。
1. 再現性について
公式のドキュメントにも記載の通り、Environment Modulesの実装はプラットフォーム特異的な部分も多く、再現性という点でcondaやcontainerを指定する場合より劣る可能性があります。
2. module: command not foundエラーについて
いくつかの条件ではmodule: command not foundというエラーとともに、モジュールが適切にロードされない場合があります。私の場合は特に、zshでsnakemakeコマンドを実行することでこの現象が起きました。zsh上ではmodule loadがちゃんと機能していたのにもかかわらすです。
いろいろ迷いましたが、これはおそらく、zsh環境で定義されていたmoduleコマンドがsnakemake側で各タスクの実行のために立ち上がるbash上に継承されないからだろうという結論に行きつきました。
moduleコマンドは実際にはシェル関数として定義されており、各種シェルスクリプト用に定義されたファイルが存在します。moduleコマンドを利用するためにはそのファイルを事前に読み込む必要があるのですが、多くの場合は.bash_profileや.bashrc、.zshrcなどのようなファイルによってログインの際に自動的に定義されるかと思います。このあたりの内容はこちらの記事が参考になります。
bashでログインし、そのままsnakemakeを起動すると、現在のシェルで定義されているmodule関数がうまいことsnakemake側で起動するbashに継承され、moduleコマンドが見つかるのだと思います。一方でzshから立ち上げるとその継承は働かず、snakemake側で起動するbashは素のbashとなり、module: command not foundエラーが発生するものと思われます。
snakemakeが起動するbashはlogin/interactive shellではないことが原因かと思います。module loadが実行される前に明示的にmoduleの初期化スクリプトをsourceする(. /etc/profile.d/modules.shといったコードを実行する)方法も考えましたが、それを実現する手段は見つかりませんでした。
簡単な解決策はsnakemakeでuse-envmodulesを利用するときはbashを使うということかと思います。(ちなみに、同じようなワークフロー言語のnextflowでも似た事象にあったことがあるのですがこちらはまた別記事で書こうと思います。)
参考
- https://modules.readthedocs.io/en/latest/
- https://snakemake.github.io/
- https://snakemake.readthedocs.io/en/stable/snakefiles/deployment.html#using-environment-modules
- https://modules.readthedocs.io/en/latest/INSTALL.html#enable-modules-in-shells
- https://github.com/snakemake/snakemake-executor-plugin-slurm/issues/63 (
module loadが実行される前に初期化スクリプト読み込みコマンドを挿入できるような機能追加をする動きに関するGithub Issues)
nextflowを-bgなしでバックグラウンド実行
個人的備忘録です。
背景と生じた問題点
nextflowをバックグラウンド実行するときは多くの場合、-bgオプションをつけて実行するかと思います。
ただ、先日シェルスクリプトで複数回nextflowを実行したいというケースにあたり、以下のようなシェルスクリプトを用意してバックグラウンド実行したところ、パイプラインがうまく走りませんでした。
スクリプトの内容
#!/bin/bash nextflow run main.nf -params-file params1.json >log1 nextflow run main.nf -params-file params2.json >log2
実行コマンド
./run.sh &
こうするとバックグラウンドプロセス./run.shはStoppedの状態になって永久に動きません。
ここで、&をつけずにシェルスクリプト自体はフォアグラウンドで実行するとなぜか正常に作動しました。(バックグラウンドで動かしたいのでこれでは意味ない。)
解決策
詳細はよくわからなかったのですが、こちらの議論によれば、-ansi-log falseまたは-quietというオプションつきで実行すれば解決するとのことで、実際、以下のようにスクリプトを変更すると動きました。
#!/bin/bash nextflow run main.nf -params-file params1.json -ansi-log false >log1 nextflow run main.nf -params-file params2.json -ansi-log false >log2
先述のリンク先の議論では-ansi-log falseまたは-quietを指定しないと入力を待つという仕様とのことのようでしたが、なぜそういう仕様なのかはよくわかりませんでした。
とりあえず問題が解決したのでよしとします。
参考
AlphaSimR使ってみた
AlphaSimRとは
こちらの論文で紹介されている、植物および動物の育種プログラムをシミュレートするためのRパッケージです。Faux et al., 2016で紹介された’AlphaSim’の後継にあたり、Rの関数を実行することで育種をシミュレートします。遺伝的な集団構造の設定、交配や表現型の取得などが関数になっていてそれらを組み合わせて一連の育種業務をシミュレートすることができるようです。
今回は初めてこのパッケージを使ってみようと思っているのでこちらにある”Introduction to AlphaSimR”を読みながら時々立ち止まりつつ試してみたいと思います。
パッケージのロード
まず、下準備としてパッケージのロードと、乱数シードの設定を行います。
library(AlphaSimR) ##> Loading required package: R6 library(ggplot2) set.seed(utf8ToInt("🐶"))
先述したIndroductionのページでは、AlphaSimRで育種プログラムのシミュレートをする上での一般的な手順として、以下の4ステップがあげられています。今回の記事でもこの手順で進めつつ、各ステップで登場する関数とオブジェクトについて深掘りしながら見ていきたいと思います。
- Creating Founder Haplotypes
- Setting simulation
- Modeling the Breeding Program
- Examining the Results
(https://cran.r-project.org/web/packages/AlphaSimR/vignettes/intro.html より引用)
1. 創始者集団のハプロタイプを構築する (Create Founder Haplotypes)
最初に登場する関数はquickHaplo()です。こちらは創始者集団のハプロタイプを定義し、MapPopというオブジェクトとして返します。この関数は完全にランダムにハプロタイプを割り当てるので、より現実的なハプロタイプを得るにはrunMacs()やnewMapPop()関数の利用が推奨されます3。
# 10染色体を持ち、各染色体に1000カ所の均等な分離するサイト(locus)を持つ仮想的な集団1000個体を生成 founderPop <- quickHaplo(nInd = 1000, nChr = 10, segSites = 1000) class(founderPop) # オプジェクトのクラス名を見る ##> [1] "MapPop" ##> attr(,"package") ##> [1] "AlphaSimR" sloop::otype(founderPop) # オブジェクトのタイプを見る ##> [1] "S4" print(founderPop) ##> An object of class "MapPop" ##> Ploidy: 2 ##> Individuals: 1000 ##> Chromosomes: 10 ##> Loci: 10000
上記の例ではオプションに、染色体数として nChr = 10
、1染色体あたりの分離するサイト(座)数として segSites = 1000
を指定していました。そのため、printの出力では Loci: 10000 (= 10 *
1000) となっています。
MapPopオブジェクトに含まれる情報の中で重要なものの1つが遺伝地図情報かと思います。遺伝地図の情報を見てみるには
founderPop@genMap
とするか、getGenMap()関数を用います。以下では後者を利用して遺伝地図情報をデータフレームとして得た結果です。
head(getGenMap(founderPop)) ##> id chr pos ##> 1 1_1 1 0.000000000 ##> 2 1_2 1 0.001001001 ##> 3 1_3 1 0.002002002 ##> 4 1_4 1 0.003003003 ##> 5 1_5 1 0.004004004 ##> 6 1_6 1 0.005005005
表で見てもイメージしづらいので、getGenMap()で得られる情報から遺伝地図を簡易的にプロットしてみたいと思います。ただし、10000
locusもあると図が見づらくなってしまうので、遺伝地図情報に関係するオプションを適宜変えながら、図を見やすくするついでに、オプションを変えたときの結果として得られる遺伝地図の変化も見ていきます。
simple_genmap_plot <- function(mappop) { getGenMap(mappop) |> ggplot(aes(chr, pos)) + geom_line() + geom_point() + ggrepel::geom_text_repel(aes(label = id)) + scale_y_reverse() } # 染色体数を3、染色体毎サイト数を3に下げる simple_genmap_plot(quickHaplo(nInd = 1, nChr = 3, segSites = 3))

# 染色体数を3、サイト数はベクトルで指定することで染色体ごとに変えられる simple_genmap_plot(quickHaplo(nInd = 1, nChr = 3, segSites = c(3, 5, 7)))

# genLenオプションで染色体の長さを変えられる(segSites同様ベクトルでの指定も可) simple_genmap_plot(quickHaplo(nInd = 1, nChr = 3, segSites = 10, genLen = 2.0))

# runMacsを使うと現実に即したより複雑な遺伝地図が構築される simple_genmap_plot(runMacs(nInd = 1, nChr = 3, segSites = 10, species = "WHEAT"))

ここで、genMapのpos列の単位について個人的に少し気になったのですが、マニュアルにその言及がある箇所を見つけられませんでした。おそらくはM(モルガン)だろうなと思いますが、あとで組換え価を計算することによって実験的にこの予想の間接的な証拠を見てみたいと思います。
2. シミュレーションの設定 (Setting Simulation Parameters)
シミュレーションを実行していく上で、形質値がどんなQTLで定義されるのか、性別はセットするのかなど、最初に設定しておくべき項目を定義することができます。そのためにはまずSimParam$new()関数でSimParamオブジェクトを生成します。ここで、変数名をSP(大文字)としておくことには大きな理由があります。AlphaSimRで使われる多くの関数は内部でグローバル変数をSPという名前で検索し利用します。Introductionでも”The
name SP should almost always be used,
“と書いてあるので、ここは従います3。
ここで注意なのが、SPという名前の変数が存在する中で、SPを上書き変更しようと思って間違えて、たとえばspという小文字の別の変数に格納した場合は以降の関数にその変更の影響を与えることができなくなってしまうことです。しかし、エラーは起きないのでこのバグに気づきづらいという状態になります。個人的にはこういう状況があるのはプログラミング上よくないような気がしますが、1スクリプトで1シミュレーションにとどめるなどしたら良いのかもしれません。
SP <- SimParam$new(founderPop) class(SP) ##> [1] "SimParam" "R6" sloop::otype(SP) ##> [1] "R6"
SimParamオブジェクトの実体はR6オブジェクトであり、Pythonといった他の多くのプログラミング言語に採用されているオブジェクト指向プログラミングの仕組みと似たものになっています。つまり、SimParamオブジェクトにはメソッドが実装されており、そのメソッドを呼び出すことで設定を実行していきます。
以下では1染色体につき、1000個の相加効果を持つQTLを設定します。また、性別については、シミュレーションの過程で集団に追加されていく順番に、システマチックに性別を割り当てるように設定しています。
SP$addTraitA(nQtlPerChr=1000) SP$setSexes("yes_sys")
SimParamクラスに実装されているメソッドは ?SimParam
で見ることができます。非常に多くのメソッドがあり、さまざまな設定が可能であることがわかります。
3. 育種業務をモデル化する (Modeling the Breeding Program)
今回の設定はこれで全て終わったので、ここで具体的な育種の過程をコードで表現していく段階になります。まずは創始者ハプロタイプと設定したパラメータから、初期育成集団を作成することになります。newPop()関数を使うことで表現します。ここで、先ほど作成したSPがコードには全く出てきませんが、裏でSPという名前の変数を読み込んでいるため、先ほどの設定はちゃんと読み込まれていることに注意です。
pop <- newPop(founderPop) class(pop) ##> [1] "Pop" ##> attr(,"package") ##> [1] "AlphaSimR" print(pop) ##> An object of class "Pop" ##> Ploidy: 2 ##> Individuals: 1000 ##> Chromosomes: 10 ##> Loci: 10000 ##> Traits: 1
newPop()関数で得られるオブジェクトはPopクラスです。シミュレーションの過程ではこのクラスのオブジェクトに対して各種操作(選抜や交配)を適用していくことになります。print(pop)の出力を見てみると、先ほどのMapPopオブジェクトの出力で見られていた情報に加え、Traitsという項目が増えていることがわかり、
先ほどのSP$addTraitA(nQtlPerChr=1000)
というコードがちゃんと機能していることがわかります。
実際にこのTraitsに関するGenetic
valueをgv()関数により取得し分布を見ると以下のようになります。
firstPopGv <- gv(pop) hist(firstPopGv, probability = TRUE) # 正規分布の密度関数も一緒にプロット curve(dnorm(x), from = min(firstPopGv), to = max(firstPopGv), add = TRUE)

SP$addTraitA()はデフォルトで平均0分散1の正規分布に従うような相加効果を与えるらしいです。
ちなみに、Additive effectによるGenetic
valueがどのように計算されているかをあえて手動で計算すると以下のようになるかと思います。遺伝子型はpullQtlGeno()で、各QTLの効果はSimParamオブジェクトのtraitsプロパティからアクセスできます。
firstPopQtlGeno <- pullQtlGeno(pop) unique(as.vector(firstPopQtlGeno)) ##> [1] 0 1 2 summary(SP$traits[[1]]@addEff) ##> Min. 1st Qu. Median Mean 3rd Qu. Max. ##> -5.272e-02 -9.382e-03 -2.498e-05 -2.858e-05 9.293e-03 4.918e-02 SP$traits[[1]]@intercept ##> [1] 0.02822522 # Additive EffectによるGenetic valueを遺伝子型と書くlocusの効果から手動で計算 firstPopGvManual <- (firstPopQtlGeno - 1) %*% SP$traits[[1]]@addEff + SP$traits[[1]]@intercept # gv()関数の結果と一致することを確認 all.equal(firstPopGv, firstPopGvManual, check.attributes = FALSE) ##> [1] TRUE
これをみると遺伝子型は0, 1, 2でコードされており、0のときはその遺伝子座の効果を引き、1のときは何もせず(中間の効果)、そして2のときは効果を足し合わせているようなイメージになっているかと思います。
それでは、実際に育種プログラム全体をコードで表現してみようと思います。以下のコードでは、狭義の遺伝率h2 = 0.2として表現型をとり(setPheno()関数)、20回の世代更新を行なっています。各世代では表現型値で上位のメス500個体とオス25個体を選抜してランダムに1000組合せの交配を行い(selectCross()関数)、後代を得ています。また、各世代での平均表現型値を取得して(meanP関数)、phenoMean変数に記録しています。
pop <- setPheno(pop, h2 = 0.2) phenoMean <- meanP(pop) for(generation in 1:20){ pop <- setPheno(pop, h2 = 0.2) pop <- selectCross(pop=pop, nFemale=500, nMale=25, use="pheno", nCrosses=1000) pop <- setPheno(pop, h2 = 0.2) phenoMean <- c(phenoMean, meanP(pop)) }
4. 結果を評価する (Examining the Results)
最後に先ほどの20世代の育種プログラムの結果を、世代を通しての平均表現型値の変遷の折れ線グラフとしてプロットしてみます。
plot(0:20, phenoMean, xlab="Generation", ylab="Mean Phenotype Value", type="l")

各世代で順調に平均表現型値が上がっていて育種の効果があることを示しています。実際の育種現場ではさらに何個もの形質を評価しながら進めていかなければならないのでこんな単純なケースはそうそう無いとは思いますが、ひとまず、単純な育種プロセスをシミュレートすることができました。
Breeder’s equation
Breeder’s equationをシミュレーションから見てみたいと思います。1世代経たときの親集団と子集団の形質平均値の差をR(引用記事中ではΔZ)、選択された集団と元の集団の形質平均値の差S(selection differential)、狭義の遺伝率をh2とするとBreeder’s equationは以下の様になります。
R = h2 * S
h2はsetPheno()関数から設定可能なので、いくつかのh2についてシミュレートしてみたいと思います。
親集団1000個体から形質値で上位10%を選抜して、ランダムに交配し後代についても1000個体得るということを5回試行し、結果を表示してみます。結果のプロットではh2をx軸にとり、R/Sの値をy軸にとって散布図をかきました。Breeder’s equationから、このプロットはy=xの直線上付近に見られると予想されます。
examine_breeders_equation <- function(h2) { pop <- newPop(founderPop) pop <- setPheno(pop, h2 = h2) n <- nInd(pop) popSel <- selectInd(pop, nInd = n %/% 10, use = "pheno") pop2 <- randCross(popSel, nCrosses = n) pop2 <- setPheno(pop2, h2 = h2) c( R = unname(meanP(pop2) - meanP(pop)), S = unname(meanP(popSel) - meanP(pop)), h2 = h2 ) } breeders_equation_result <- purrr::map( seq(0.1, 1.0, 0.2), ~as.data.frame(t(replicate(5, examine_breeders_equation(.x)))) ) |> dplyr::bind_rows() breeders_equation_result |> ggplot(aes(h2, R/S)) + geom_point() + geom_abline(slope = 1, intercept = 0) + coord_equal()

実際y=x付近にプロットされました。
世代を超えたIBDの追跡(組換えの可視化)
AlphaSimRではSP$setTrackRec(TRUE)とすることで組換えイベントを保持してシミュレーションしてくれます。これを行うと、pullIbdHaplo()関数で集団のハプロタイプをIBD(Identidy
by
descent)ベースで見ることができます。これは創始者集団の染色体断片がどのように後代に遺伝するかを見ることができるということです。そこで、簡単なグフラフィカルジェノタイプ的なプロットをする関数を作って実際に集団のいくつかの個体についてのIBDハプロタイプを見てみたいと思います。
描画を簡単にするため創始者集団は2個体の二倍体で、1染色体のみを持つとします。すると創始者集団にはハプロタイプとして合計4種類を含むことになり、これらを4色で色分けしてプロットします。また、データ数が多くなるとグラフが見にくくなるので、各世代10個体のみを得ることにしました。
simple_ibd_haplotype_plot <- function(pop, map) { pullIbdHaplo(pop) |> as.data.frame() |> tibble::rownames_to_column("haplo_id") |> tidyr::pivot_longer(-haplo_id, values_to = "haplotype") |> dplyr::mutate( haplotype = factor(haplotype) ) |> dplyr::full_join(map, by = c("name" = "id")) |> ggplot(aes(pos, haplo_id)) + geom_raster(aes(fill = haplotype)) + theme(axis.text.x = element_blank()) } founderPop <- quickHaplo(2, nChr = 1, segSites = 100) map <- getGenMap(founderPop) SP <- SimParam$new(founderPop) SP$setTrackRec(TRUE) pop <- newPop(founderPop) simple_ibd_haplotype_plot(pop, map) # 最初の集団

それでは、上記4つに色分けされたハプロタイプ(染色体)がどう後代に遺伝するかを追ってみます。
S1世代
組換えが起こり、色分けにスイッチングしている部分が見られます。二倍体が10個体なので、合計20本分のデータがプロットに見られます。
pop <- makeCross(pop, crossPlan = matrix(c(1, 2), ncol = 2), nProgeny = 10) simple_ibd_haplotype_plot(pop, map) # 第1世代

S2世代
pop <- randCross(pop, nCrosses = 10) simple_ibd_haplotype_plot(pop, map) # 第2世代

S10世代
世代を経るごとに連鎖ブロックが途切れ途切れになっている様子が見えるかと思います。また、集団サイズを10と小さく取っているからか2番目のハプロタイプがかなり除かれていることが見て取れます。
for (i in 1:8) { pop <- randCross(pop, nCrosses = 10) } simple_ibd_haplotype_plot(pop, map) # 第10世代

遺伝距離と組換え価の関係
先ほど、getGenMap()で得られるpos列の単位について、おそらくM(モルガン)であろうという話をしましたが、それを間接的に裏付けるデータを取ってみようと思います。具体的には親を1個体を用意して、その自殖第1世代(S1)1000個体を得るということをシミュレーションしてみます。このとき、親、およびS1のハプロタイプはpullSegSiteHaplo()で完全なものが得られるため、簡単に組換え価が計算できます。具体的な計算はコードを見ていただくとして、実際に組換え価とposの値の差の関係をプロットしてみます。
founderPop <- runMacs(nInd = 1, nChr = 1, segSites = 20, species = "MAIZE") pos <- getGenMap(founderPop)$pos SP <- SimParam$new(founderPop) # indexが1の個体を親として新しいPopオブジェクト作成 parent <- newPop(founderPop[1]) haplo_p <- pullSegSiteHaplo(parent) haplo_p_sub <- haplo_p[, haplo_p[1, ] != haplo_p[2, ]] # 後代で分離するサイトのみ保持 # 親個体を自殖して1000個体を得る s1 <- self(parent, nProgeny = 1000) haplo_s1_sub <- pullSegSiteHaplo(s1)[, haplo_p[1, ] != haplo_p[2, ]] # 各サイト間の遺伝距離(pos列の差分)と組換え価を計算する n <- ncol(haplo_p_sub) recomb_rate <- NULL pos_diff <- NULL for (i in 1:(n-1)) { for (j in (i+1):n) { match_to_haplo1 <- apply(haplo_s1_sub[, c(i, j)], 1, function(x) all(x == haplo_p_sub[1, c(i, j)])) match_to_haplo2 <- apply(haplo_s1_sub[, c(i, j)], 1, function(x) all(x == haplo_p_sub[2, c(i, j)])) recomb_rate <- c(recomb_rate, mean(!(match_to_haplo1 | match_to_haplo2))) pos_diff <- c(pos_diff, abs(pos[j] - pos[i])) } } plot(pos_diff, recomb_rate) curve((1-exp(-2 * x))/2, from = min(pos_diff), to = max(pos_diff), add = TRUE, col = "blue") curve(tanh(2 * x)/2, from = min(pos_diff), to = max(pos_diff), add = TRUE, col = "red") legend("bottomright", c("Haldane", "Kosambi"), col = c("blue", "red"), lty =c(1, 1))

するとこれらの関係は綺麗にKosambi mapping function上に乗ってきます。このことから、おそらくposはM単位の値であると考えられるかと思います。
あえてシミュレーションパラメータにSPという名前をつけない例
最後に、AlphaSimRでは独特の作法でSimParamオブジェクトにはSPという変数名をつけるということが推奨されています。ただ、これは一般的なプログラミングにおいては普通ではないので、なんとなく違和感を感じるという方のために、SPという変数名を使わない場合のコードを以下に置いておきます。
具体的には各関数にあるsimParam引数にSimParamオブジェクトを格納した変数を与えてやることで可能になります。以下の例では大文字のSP(推奨の変数名)の代わりに小文字のspという変数名を用いてみています。ちなみにこちらの例をもとにしています。
rm(list = ls()) # 先ほど定義したSPとその他もろもろをを消しておく founderPop <- quickHaplo(nInd=1000, nChr=10, segSites=1000) sp <- SimParam$new(founderPop) sp$addTraitA(nQtlPerChr=1000) sp$setSexes("yes_sys") #pop <- newPop(founderPop) # SPという変数がない場合、これではerrorが発生 pop <- newPop(founderPop, simParam = sp) genMean <- meanG(pop) for(generation in 1:20){ pop = selectCross(pop=pop, nFemale=500, nMale=25, use="gv", nCrosses=1000, simParam = sp) genMean <- c(genMean, meanG(pop)) } # Examining the Results plot(0:20, genMean, xlab="Generation", ylab="Mean Genetic Value", type="l")

参考文献
- Gaynor, R. Chris, Gregor Gorjanc, and John M. Hickey. 2021. AlphaSimR: an R package for breeding program simulations. G3 Gene|Genomes|Genetics 11(2):jkaa017. https://doi.org/10.1093/g3journal/jkaa017.
- Faux, A.-M., Gorjanc, G., Gaynor, R.C., Battagin, M., Edwards, S.M., Wilson, D.L., Hearne, S.J., Gonen, S. and Hickey, J.M. (2016), AlphaSim: Software for Breeding Program Simulation. The Plant Genome, 9: plantgenome2016.02.0013. https://doi.org/10.3835/plantgenome2016.02.0013
- http://cran.r-project.org/web/packages/AlphaSimR/vignettes/intro.html
- https://www.nature.com/scitable/knowledge/library/the-breeder-s-equation-24204828/
- https://en.wikipedia.org/wiki/Genetic_map_function
ランダムになんかロゴみたいなもの作る
ここ最近、輪郭形状解析の話をブログにまとめている中で、ランダムな形状の生成を試行していておもしろい模様生成の方法をたまたま見つけたのでその内容をメモがてら記事に残しておこうと思います。
具体的なコードと生成された模様は以下。ことの顛末としては、楕円フーリエ解析における係数からの輪郭再構築の過程を模してランダムな形状生成を試行錯誤していたのですが、そのとき、係数と
を0にすると左右対称のパターンが生成され、なんかロゴマークみたいでおもしろいと思って記事にしました。関数の
order引数で重ね合わせる波の周波数の数を設定します。係数と
を生成する乱数を正規分布ではなくて別の分布を使えばまた違ったパターンが見えるかもしれません。以下では
numpy.random.normalとnumpy.random.choiceを利用した場合を記載しています。
numpy.random.normal利用
import numpy as np import matplotlib.pyplot as plt def generate_random_contour(order=5): t = np.linspace(0, 1, num=1000) phi = (2 * np.pi * t) * np.arange(1, order+1).reshape((order, 1)) b = np.random.normal(size=order) c = np.random.normal(size=order) x = np.dot(b, np.sin(phi)) y = np.dot(c, np.cos(phi)) return x, y for order in [2, 3, 4, 5, 10, 20]: fig, axes = plt.subplots(10, 10, figsize=(15, 15)) for ax in axes.ravel(): x, y = generate_random_contour(order) ax.plot(x, y) ax.set_aspect(1) ax.axis('off') fig.suptitle(f"order={order}") plt.show()






numpy.random.choice利用
import numpy as np import matplotlib.pyplot as plt def generate_random_contour(order=5): t = np.linspace(0, 1, num=1000) phi = (2 * np.pi * t) * np.arange(1, order+1).reshape((order, 1)) b = np.random.choice([0, 0.1, 0.2, 1], size=order, p=[0.4, 0.1, 0.2, 0.3]) c = np.random.choice([0, 0.1, 0.2, 1], size=order, p=[0.4, 0.1, 0.2, 0.3]) x = np.dot(b, np.sin(phi)) y = np.dot(c, np.cos(phi)) return x, y for order in [2, 3, 4, 5, 10, 20]: fig, axes = plt.subplots(10, 10, figsize=(15, 15)) for ax in axes.ravel(): x, y = generate_random_contour(order) ax.plot(x, y) ax.set_aspect(1) ax.axis('off') fig.suptitle(f"order={order}") plt.show()






楕円フーリエ解析の前の補正(線対称な図形の場合)
- 必要なパッケージのインポート
- テスト用画像生成
- 輪郭抽出
- 中心座標を揃える
- サイズを揃える
- 向きを揃える
- 得られた回転角情報をもとに輪郭の向きを揃える
- 開始点と点列の向きをそろえる
- 本番の楕円フーリエ解析
- 参考文献
前回の記事で楕円フーリエ解析における標準化は結果の解釈を大きく変える可能性があり、きちんと考える必要があることを紹介しました。 また、その中で輪郭に前処理を行い向きや輪郭の開始点を調整しておくと良いこともわかりました。
しかし、前回はシミュレーションの仮定から輪郭の向きや開始点の補正を逆算的に行なっており、この方法は実践的ではありません。 そこで今回はより実践に近い形でのシミュレーションもしていこうと思います。輪郭の向きや開始点の正解がわからないという状況を再現して輪郭補正をやってみたいと思います。
また、今回重要な仮定として、輪郭が線対称となっていることという条件を設けています。この事実から、その軸を元に向きを補正して楕円フーリエ解析を行うという流れで解析例を実験してみたいと思います。実践の中では線対称でない図形を対象にする場合も多いかと思いますが、そのような場合でも、何が図形の向きを揃えるという意味で特徴的な軸がある場合は参考になるかもしれません。
必要なパッケージのインポート
import numpy as np import pandas as pd import cv2 import matplotlib.pyplot as plt import seaborn as sns from pyefd import elliptic_fourier_descriptors, normalize_efd, reconstruct_contour from sklearn.decomposition import PCA from scipy.spatial import KDTree from scipy.optimize import minimize
テスト用画像生成
今回は最初から輪郭の点列が座標として与えられているという状況の一歩前から実験用のテストデータを作成していこうと思います。輪郭の点列データが得られる前は、対象となる物体と背景を識別したニ値画像を持っている状況が多いと思います。そこで今回は前回と同じ輪郭データに対し、ランダムに回転させ、(回転角 )、ランダムに拡大縮小・平行移動し、ランダムな小さいノイズを加えています。この輪郭データを元に
cv2.fillPoly関数を用いて二値画像を作成しています。
あとで図形の回転角psiを統一するという処理が目標の1つになるので、正解の図形の回転角も変数true_psiに保持しておきます。
np.random.seed(ord("🐱")) images = np.zeros((100, 500, 500), np.uint8) i = 0 true_psi = [] for a in np.linspace(-1, 1, 10): for d in np.exp(np.linspace(np.log(1/2), np.log(2), 10)): # テスト用輪郭の生成 theta = np.linspace(0, 2*np.pi, 100)[:-1] x = np.cos(theta) + a * np.cos(theta*2) y = d * np.sin(theta) # ランダムに回転 psi = np.random.uniform(0, np.pi) rot = np.array([[np.cos(psi), -np.sin(psi)], [np.sin(psi), np.cos(psi)]]) x, y = rot @ np.stack((x, y), axis=0) true_psi.append(psi) # 一旦面積を揃えてから平均100の拡大に関する係数をかける tmp = np.stack([x, y], axis=1).reshape((-1, 1, 2)) area = cv2.contourArea(tmp.astype(np.float32)) s = np.random.normal(loc=100, scale=10) x *= s / np.sqrt(area) y *= s / np.sqrt(area) # ランダムなノイズを加える x += np.random.normal(size=len(x)) y += np.random.normal(size=len(y)) # 余白10px含め画像500x500に入らない場合はサイズダウン max_side_size = max(np.ptp(x), np.ptp(y)) if max_side_size > 480: x *= 480 / max_side_size y *= 480 / max_side_size # 余白10px除いた画像中央480x480の領域にランダムに配置されるようにする # または中心座標が200~300に入るようにする x += np.random.uniform(max(10 - x.min(), 200 - x.mean()), min(490 - x.max(), 300 - x.mean())) y += np.random.uniform(max(10 - y.min(), 200 - y.mean()), min(490 - y.max(), 300 - y.mean())) # 輪郭をもとに画像に塗りつぶし pts = np.stack((x, y), axis=1).reshape(-1, 1, 2).round().astype(np.int32) cv2.fillPoly(images[i], [pts], 255, 8, 0) i += 1 # テスト画像のいくつかを抜粋 fig, axes = plt.subplots(1, 5, figsize=(10, 2)) axes = axes.ravel() for i, img in enumerate(images[::20]): ax = axes[i] ax.imshow(img, cmap="gray") plt.show()

輪郭抽出
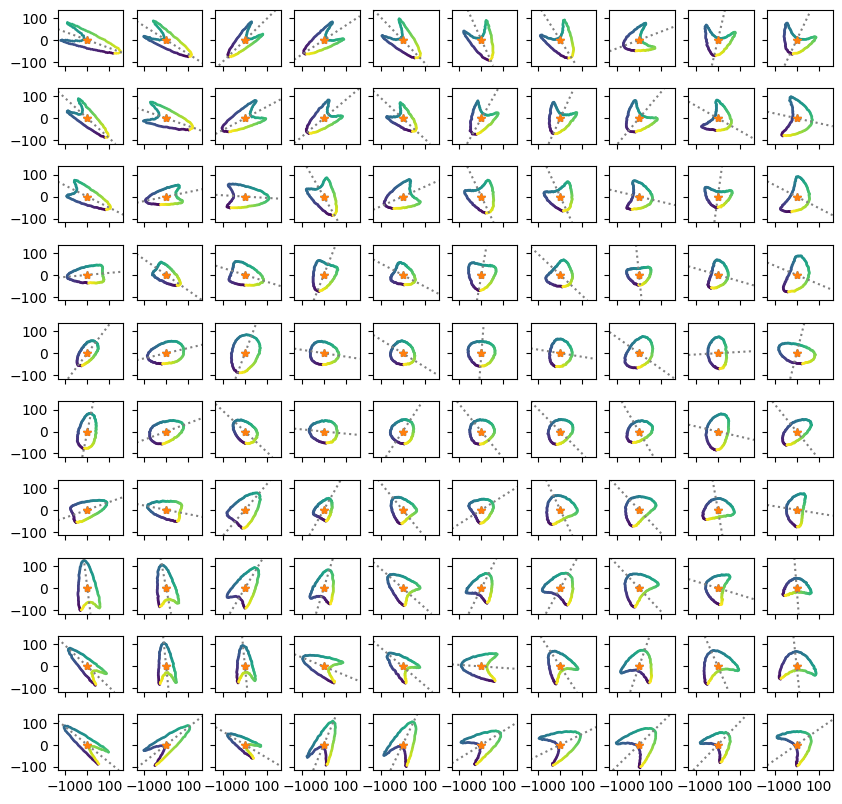
輪郭抽出の一般的な方法に従いcv2.findContours関数を用いた輪郭検出を行います。描画用の関数plot_contourも用意しておきます。これはtrue_psiには真の回転角、pred_psiには予測された回転角をとり、それぞれ角度の軸を描画します。
def plot_contour(contours, true_psi, pred_psi=None): fig, axes = plt.subplots(10, 10, sharex=True, sharey=True, figsize=(10, 10)) axes = axes.ravel() # 輪郭描画 for ax, cnt in zip(axes, contours): x, y = cnt[:, 0, :].T ax.scatter(x, y, c=range(len(x)), s=1) # 輪郭中心座標の計算 centers = np.array(contours).mean(axis=(1, 2)) # 真の傾き描画 for ax, psi, center in zip(axes, true_psi, centers): ax.axline(center, slope=np.tan(psi), linestyle="dotted", color="gray") # 予測された傾き描画 if pred_psi is not None: for ax, psi, center in zip(axes, pred_psi, centers): ax.axline(center, slope=np.tan(psi), linestyle="dashed", color="orange") for ax in axes: ax.set_aspect(1) #ax.axis('off') return fig, axes
contours = [] centers = [] for img in images: cnts, _ = cv2.findContours(img, cv2.RETR_EXTERNAL, cv2.CHAIN_APPROX_NONE) assert len(cnts) == 1 #contourSamping関数が使えるのであれば等間隔に座標を再サンプリング cnt = cv2.ximgproc.contourSampling(cnts[0], nbElt=100) contours.append(cnt) plot_contour(contours, true_psi) plt.show()

中心座標を揃える
まずは、最も簡単に楕円フーリエ解析で標準化を用いない際に重要な前処理となる中心座標(重心)を揃えることを行いたいと思います。原理的にはどこに揃えてもいいような気はしますが、わかりやすいので原点に合わせる処理を行います。この処理は具体的には各座標の平均をすべての点の座標の値から引いてあげれば良いです。図では原点を青色、輪郭の中心をオレンジ色で描画しており、それらが重なっていることがわかるかと思います。
contours_centered = [cnt - cnt.mean(axis=(0, 1)) for cnt in contours] fig, axes = plot_contour(contours_centered, true_psi) for ax, cnt in zip(axes, contours_centered): ax.plot(0, 0, '*') ax.plot(cnt[:, 0, 0].mean(), cnt[:, 0, 1].mean(), '*') plt.show()
サイズを揃える
楕円フーリエ解析における標準化では第一調和楕円の長軸が一定になるように標準化がされていますが、今回はその情報を用いると細長い輪郭と扁平な輪郭で差が出てしまうことが前回の記事からも予想される懸念としてあるので、面積が1となるような前処理(=面積の平方根で座標の値を割る)を行いたいと思います。この点に関しても、各ケースで揃えるべき部分は変わるかと思います。例えば今回の場合でも矢じりの長さを一定にした場合の輪郭の特徴の差を見たいというケースでは別の前処理が必要になってくるかと思います。こう考えていくと形を揃えるといったことは言葉では簡単に言えますが、なかなか難しいことだと感じます。
contours_resized = [] for cnt in contours_centered: area = cv2.contourArea(cnt) contours_resized.append(cnt / np.float32(np.sqrt(area))) plot_contour(contours_resized, true_psi) plt.show()

向きを揃える
ここで一番の難関、向きを揃えることを目指していこうと思います。もちろんtrue_psiに格納されている情報は実践の場では未知の情報のため使えません。
今回は輪郭の特性として、「線対称である」ということを利用し、対象軸をまず検出して、その角度をもとに輪郭の軸を揃えていきます。
対称性を測るクラスを定義
輪郭の対称性を見つける上で対称性とは何かを定義してあげる必要があります。ここでは輪郭をある軸について線対称になるように変換した輪郭を作り、それと元の輪郭の差異が小さいとき、その軸は対称軸であるだろうと考えます。ここで2つの輪郭の差異はscipy.spatial.KDTreeを用いて測ります。energyメソッドを回転角psiを引数にして呼ぶとその図形中心を通る、x軸とのなす角がpsiの線で対象な図形との差異を計算します。この値が小さいほど、そのpsiは対称軸の回転角らしいと言える指標になります。
class ReflectionSymmetry(): def __init__(self, contour): contour = np.squeeze(contour) self.contour = contour - contour.mean(axis=0) self.tree = KDTree(contour) def reflect(self, psi): n = np.array([-np.sin(psi), np.cos(psi)]) # 法線ベクトル dist = self.contour @ n return self.contour - 2 * dist[:, None] * n def energy(self, psi): reflect_contour = self.reflect(psi) dists, _ = self.tree.query(reflect_contour, k=1) return dists.mean()
輪郭に対してのPCA
広く使われている輪郭の軸の検出で、主成分分析(PCA)があります。点列のx座標とy座標に対してPCAを適用した際の主成分軸が対称線に重なることが多いためです。これはどんなケースでも利用できる方法ではないので、一旦、今回のケースでどうなるかをみてみます。主成分軸が2つ得られるので、そのうちどちらを採用するかは先ほどのenergyメソッドの値が低い方にします。推定された軸をオレンジ色で描画します。
def pred_psi_by_pca(contour): pca = PCA(n_components=2).fit(contour.reshape((-1, 2))) psi1 = np.arctan2(pca.components_[0, 1], pca.components_[0, 0]) psi2 = np.arctan2(pca.components_[1, 1], pca.components_[1, 0]) rs = ReflectionSymmetry(contour) if rs.energy(psi1) < rs.energy(psi2): psi = psi1 else: psi = psi2 return psi pred_psi_pca = [pred_psi_by_pca(cnt) for cnt in contours_resized] fig, axes = plot_contour(contours_resized, true_psi, pred_psi_pca) plt.show()

概ねうまく回転角を推定できていますが、一部大きく外しているものもあります(真の軸(灰色点線)と予測された軸(オレンジ点線)がズレる)。推定が外れるものは比較的縦長でも横長でもない中間的な形状を持っている傾向があります。残念ながら今回は使えなさそう(一部マニュアルで回転角を設定するといった対応が取れる場合はありかも)なので、別の方法を試します。
2次のモーメントを利用
原理的にはPCAと同じ方法ですが、opencvのmoments関数を使った場合も試してみたいと思います。ここで、回転角はこちらのサイトを参考にしました。こちらも推定された回転角がちょうど90どズレる可能性(横長の矢じりの場合)があるので先ほどのPCAと同様の基準で推定された角度とその+90度の2つから1つを選択します。
def pred_psi_by_moment(contour): m = cv2.moments(contour) mu11, mu20, mu02 = m['mu11'], m['mu20'], m['mu02'] psi1 = 0.5 * np.arctan2(2 * mu11, mu20 - mu02) psi2 = (psi1 + (np.pi/2)) % (np.pi*2) rs = ReflectionSymmetry(contour) if rs.energy(psi1) < rs.energy(psi2): psi = psi1 else: psi = psi2 return psi pred_psi_mmn = [pred_psi_by_moment(cnt) for cnt in contours_resized] fig, axes = plot_contour(contours_resized, true_psi, pred_psi_mmn) plt.show()

原理的にはPCAと同じためか、PCAと同様にいくつかの輪郭で推定がうまくいっていないことがわかりました。
EFAの第一調和楕円の軸を利用
前回の楕円フーリエ解析の正規化の記事でも第一調和楕円の長軸の向きを合わせるといった標準化はうまく機能しないことがわかっています。しかし、短軸であれば上手い推定ができているかもしれません。そこで、長軸、短軸のうちどちらが良いかを先ほどと同様にして選択したときにどうなるかもみてみます。第一調和楕円の長軸の回転角はpyefd.normalize_efdで計算可能なのでそちらを利用します。
def pred_psi_by_efa(contour): contour = np.squeeze(contour) efd = elliptic_fourier_descriptors(contour) _, (_, psi1, _) = normalize_efd(efd, return_transformation=True) psi2 = psi1 + (np.pi/2) # 第一調和楕円の短軸の方 rs = ReflectionSymmetry(contour) if rs.energy(psi1) < rs.energy(psi2): psi = psi1 else: psi = psi2 return psi pred_psi_efa = [pred_psi_by_efa(cnt) for cnt in contours_resized] fig, axes = plot_contour(contours_resized, true_psi, pred_psi_efa) plt.show()

残念ながらこれもいくつかの輪郭で推定がうまくいきませんでした。
総当たり法
最終手段、360度を100等分して、そのうち最も対称線らしいものを対称軸として推定します。解像度は3.6度が限界ということになりますが、ここは計算量との兼ね合いで増やすこともできますし、最終的には最適化アルゴリズム(Nelder-Mead法など)を使っても良いと思います。
def pred_psi_by_bruteforce(contour): rs = ReflectionSymmetry(contour) _, psi = min([(rs.energy(psi), psi) for psi in np.linspace(-np.pi/2, np.pi/2, 100)]) return psi pred_psi_bf = [pred_psi_by_bruteforce(cnt.copy()) for cnt in contours_resized] plot_contour(contours_resized, true_psi, pred_psi_bf) plt.plot()

結局これが一番うまくいっているのでこの方法で得られた推定値をもとに図形を逆に回転させ、向きを揃えていきたいと思います。
得られた回転角情報をもとに輪郭の向きを揃える
軸を揃える
先ほど総当たり法で求めた角度と反対方向に回転させるために、回転行列を座標値にかけることで輪郭を回転させます。
contours_rotated = [] for psi, cnt in zip(pred_psi_bf, contours_resized): cnt = np.squeeze(cnt) M = np.array([ [np.cos(psi), np.sin(psi)], [-np.sin(psi), np.cos(psi)], ]) cnt = (M @ cnt.T).T.reshape(-1, 1, 2) contours_rotated.append(cnt) true_psi_rotated = np.array(true_psi) - np.array(pred_psi_bf) plot_contour(contours_rotated, true_psi_rotated) plt.show()

向きをそろえる
まだ矢じりの尖っている方とそうでない方が逆のものも存在するので、それらについて、y軸の分散の値を参考に逆転させます(y軸の分散は尖っている方が小さいという印象のため)。
for cnt in contours_rotated: left = cnt[cnt[:, 0, 0] < 0, 0, 1] right = cnt[cnt[:, 0, 0] > 0, 0, 1] if np.var(left) < np.var(right): cnt[:, 0, 0] *= -1 fig, axes = plot_contour(contours_rotated, true_psi_rotated) plt.show()

開始点と点列の向きをそろえる
最後に、点列の開始点、向き(時計回りか反時計回りか)がを揃えます。具体的にはx軸が最大の点(=矢じりの先端)を開始点になるように、np.rollで要素をずらし、点列の前半と点列の後半のy軸の平均の大小から反時計回りかどうかを検証し、反時計回りになるようにします(反時計回りの場合は先に矢じりの左半分(xy平面上ではy軸の正の方向)に向かうので、y座標の値は前半の方が大きいだろうという予想のもと)。
contours_shifted = [] for cnt in contours_rotated: new_cnt = np.roll(cnt, shift=-cnt[:, 0, 0].argmax(), axis=0) n = cnt.shape[0] former = new_cnt[:n//2, 0, 1] latter = new_cnt[n//2:, 0, 1] if former.mean() < latter.mean(): new_cnt = np.array(list(reversed(new_cnt))) contours_shifted.append(new_cnt) fig, axes = plot_contour(contours_shifted, np.array(true_psi) - np.array(pred_psi_mmn)) plt.show()

contours_final = contours_shifted
本番の楕円フーリエ解析
今回もktchを参考にした楕円フーリエ解析を行います。(https://doc.ktch.dev/notebooks/outline/elliptic_Fourier_analysis.html)。また、今回はShape on R(http://lbm.ab.a.u-tokyo.ac.jp/~iwata/software/shape_r/)のような可視化も行ってみました。これは楕円フーリエ記述子の値を主成分ごとに分けて一個ずつ変化させていった時にどのような輪郭が得られるかを重ねて描画するものです。
coefs = [elliptic_fourier_descriptors(cnt[:, 0, :]).flatten() for cnt in contours_final] pca = PCA(n_components=5) coefs_transformed = pca.fit_transform(coefs) plt.bar(range(5), pca.explained_variance_ratio_) plt.show()

まずは楕円フーリエ記述子で記述される今回の解析対象の輪郭はおおよそ2~3主成分で説明可能なことがわかります。 それでは、ktchのNotebookにあるようなMorphospaceの描画と、Shape on Rにあるような各主成分が説明する輪郭の変動の描画を行ってみます。
def get_pc_scores_for_morphospace(ax, num = 5): xrange = np.linspace(ax.get_xlim()[0], ax.get_xlim()[1],num) yrange = np.linspace(ax.get_ylim()[0], ax.get_ylim()[1],num) return xrange, yrange # 参考:https://doc.ktch.dev/notebooks/outline/elliptic_Fourier_analysis.html def plot_recon_morphs(pca, fig, ax, n_PCs_xy=[1, 2], morph_num=5, morph_scale=0.8, morph_color="gray", morph_alpha=0.7): pc_scores_h, pc_scores_v = get_pc_scores_for_morphospace(ax, morph_num+2) pc_scores_h, pc_scores_v = pc_scores_h[1:-1], pc_scores_v[1:-1] for pc_score_h in pc_scores_h: for pc_score_v in pc_scores_v: pc_score = np.zeros(pca.n_components_) n_PC_h, n_PC_v = n_PCs_xy pc_score[n_PC_h-1] = pc_score_h pc_score[n_PC_v-1] = pc_score_v arr_coef = pca.inverse_transform([pc_score]) ax_width = ax.get_window_extent().width fig_width = fig.get_window_extent().width fig_height = fig.get_window_extent().height morph_size = morph_scale*ax_width/(fig_width*morph_num) loc = ax.transData.transform((pc_score_h, pc_score_v)) axins = fig.add_axes([loc[0]/fig_width-morph_size/2, loc[1]/fig_height-morph_size/2, morph_size, morph_size], anchor='C') coords = reconstruct_contour(arr_coef[0].reshape(-1, 4)) x = coords[:,0] y = coords[:,1] axins.plot(x.astype(float),y.astype(float),color=morph_color, alpha = morph_alpha) axins.axis('equal') axins.axis('off') # 参考:http://lbm.ab.a.u-tokyo.ac.jp/~iwata/software/shape_r/ def plot_pca_recon(pca, i, unit, normalize=False, ax=None): pcscores = np.zeros((3, pca.n_components_)) pcscores[:, i] = np.array([-1, 0, 1]) * unit * np.sqrt(pca.explained_variance_[i]) coeffs = pca.inverse_transform(pcscores) coeffs = coeffs.reshape((3, -1, 4)) contours = [reconstruct_contour(coeff) for coeff in coeffs] ax.plot(contours[0][:, 0], contours[0][:, 1], 'C0--') ax.plot(contours[1][:, 0], contours[1][:, 1], 'C2') ax.plot(contours[2][:, 0], contours[2][:, 1], 'C3--') ax.set( aspect=1, title=f"PC{i+1}" ) return ax
import pandas as pd df_pca = pd.DataFrame(coefs_transformed, columns=[f"PC{i+1}" for i in range(5)]) fig, ax = plt.subplots() sns.scatterplot(data=df_pca, x="PC1", y="PC2", ax = ax, legend = True) plot_recon_morphs(pca, fig=fig, ax=ax) plt.show() fig, axes = plt.subplots(1, 3, figsize=(9, 3)) for i in range(3): plot_pca_recon(pca, i, 1, normalize=False, ax=axes[i])


前回の記事と同様にいい感じで矢じりの扁平/細長と尻部分の窪みの大小が主成分として抽出できたのではないでしょうか。これで、楕円フーリエ解析で関数備え付けの標準化(Normalization)が機能しない場合でもこのような前処理を行なっていくことで解析が可能であるという道筋が見えてきました。向きの統一はかなり難しい点で対応方法もケースバイケースだと思うので、最近ではAIの力を使うなりしたいところだと思いました(実際今回の記事を書く上でも生成AIに相談していたりします)。