この間会社の先輩から「IT農家のラズパイ製ディープ・ラーニング・カメラ」(2020、小池誠)という本を貸していただいたので読んでみました。その中でディープラーニングの応用例として枝豆の莢の画像を2粒莢と3粒莢に分けるというタスクが紹介されていました。このデータセットについては親切にGitHub上に公開されています。今回はこれをディープラーニングではない方法で分類できないか実験してみました。
使用言語はPythonで以下のモジュール・関数群をインポートしています。
このインポートの様子からもわかるように今回はランダムフォレストでの分類を試みます。
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from skimage.measure import label, regionprops
from skimage.filters import threshold_otsu
from skimage.morphology import opening, square
from sklearn.ensemble import RandomForestClassifier
from sklearn import metrics
データダウンロード・整形
まずはデータセットをGitHubからダウンロードして整形するところからです。
以下のようにして、GitHubにあるスクリプトを参考に多少Numpy配列のShapeを変更してデータセットを構築します。
このデータセットはすでに学習用と評価用に分けられているのでそれをそのまま用います。
!wget "https://github.com/workpiles/soybeans_sorter/raw/master/soybeans_gray.npz" -O soybeans_dataset.npz -q
IMAGE_HEIGHT = 48
IMAGE_WIDTH = 64
def load_data():
data = np.load("soybeans_dataset.npz")
x_train = data['x_train'].astype('float32') / 255.
x_test = data['x_test'].astype('float32') / 255.
x_train = x_train.reshape([-1, IMAGE_HEIGHT, IMAGE_WIDTH])
x_test = x_test.reshape([-1, IMAGE_HEIGHT, IMAGE_WIDTH])
y_train = data['y_train']
y_test = data['y_test']
return x_train, y_train, x_test, y_test
X_train, y_train, X_test, y_test = load_data()
print(X_train.shape, y_train.shape, X_test.shape, y_test.shape)
(600, 48, 64) (600,) (580, 48, 64) (580,)
特徴量抽出
ディープラーニングでは画像と教師データを入力すると自動で特徴量の抽出を学習してくれますが、今回はそれを用いないので自分で特徴量を抽出する必要があります。
個人的に形を見れば分類できそうに感じたので形に関連する特徴量を画像から抽出するコードを書いていきます。
まずは画像における枝豆の領域をマスク画像として抽出したいので大津法による閾値の計算を行い、画像を2値化します。
example_image = X_train[0]
thre = threshold_otsu(example_image)
mask = opening(example_image < thre, square(3))
fig, ax = plt.subplots(1, 2, figsize=(6, 4))
ax[0].imshow(example_image, cmap="gray")
ax[0].set_title("raw image")
ax[1].imshow(mask, cmap="gray")
ax[1].set_title("binary image")
plt.show()

枝豆のマスク画像を作成できたので、 skimage.measure の regionprops 関数でマスク画像に含まれる連続した白い部分ごとに抽出することができる各種特徴量について計算します。
regions = regionprops(label_image=label(mask))
prop = sorted(regions, key=lambda x:x.area, reverse=True)[0]
上記コードによって prop という変数に計算された特徴量が格納されます。
形を定量する特徴量としてHu momentsというものがあるのでこれを用います。これは図形が回転したり拡大縮小しても変化しない特徴量で形を数値化していると考えることができると思います。Hu momentsは一般に7次元のベクトルとして表されます。
また、画像取得時のカメラと対象の距離が固定されているとのことなので、枝豆の面積や長さ等の特徴量も利用できるのではと思ったのでこれらも含めたいと思います(本では2粒莢と3粒莢で長さが等しいものもあると紹介されていますが)。
feature_names = [
"hu moments 1",
"hu moments 2",
"hu moments 3",
"hu moments 4",
"hu moments 5",
"hu moments 6",
"hu moments 7",
"perimeter",
"area",
"major axis length",
"eccentricity",
]
print(f"hu moments: {prop.moments_hu}")
print(f"perimeter: {prop.perimeter}")
print(f"area: {prop.area}")
print(f"major axis length: {prop.major_axis_length}")
print(f"eccentricity: {prop.eccentricity}")
hu moments: [ 3.39570803e-01 8.53334557e-02 2.62741796e-03 4.25716946e-04
1.16743483e-07 -1.29062051e-05 -4.34843423e-07]
perimeter: 127.25483399593904
area: 647
major axis length: 57.180643020830146
eccentricity: 0.9617073387929369
これらの特徴量を抽出しnumpyのベクトルとして返す関数を用意します
def extract_features(img):
thre = threshold_otsu(img)
mask = opening(img < thre, square(3))
regions = regionprops(label_image=label(mask), intensity_image=img)
prop = sorted(regions, key=lambda x:x.area, reverse=True)[0]
features = np.concatenate((prop.moments_hu, [prop.perimeter, prop.area, prop.major_axis_length, prop.eccentricity]))
return features
この関数を用いて X_train および X_test からデータ数*特徴量数の行列( new_X_train 、 new_X_test )を作成します。
new_X_train = np.array([extract_features(x) for x in X_train])
new_X_test = np.array([extract_features(x) for x in X_test])
print(new_X_train.shape)
print(new_X_test.shape)
(600, 11)
(580, 11)
ランダムフォレストによる学習
それではいよいよランダムフォレストによる学習を実行します。今回は木の数を200に設定しました。
forest = RandomForestClassifier(n_estimators=200, random_state=0)
forest.fit(new_X_train, y_train)
y_pred = forest.predict(new_X_test)
print(f"accuracy: {metrics.accuracy_score(y_test, y_pred)}, F1: {metrics.f1_score(y_test, y_pred)}")
accuracy: 0.9913793103448276, F1: 0.9914236706689538
無事正答率、F値ともに0.99の精度で評価用データセットを分類することができました。
各特徴量の重要度
ランダムフォレストの良い点として各特徴量の予測に対する重要度を計算することができます。そこでここでも重要度を算出してプロットしてみます。
importances = forest.feature_importances_
std = np.std([tree.feature_importances_ for tree in forest.estimators_], axis=0)
forest_importances = pd.Series(importances, index=feature_names)
fig, ax = plt.subplots(figsize=(6, 4))
forest_importances.plot.bar(yerr=std, ax=ax)
ax.set_title("Feature importances using MDI")
ax.set_ylabel("Mean decrease in impurity")
fig.tight_layout()
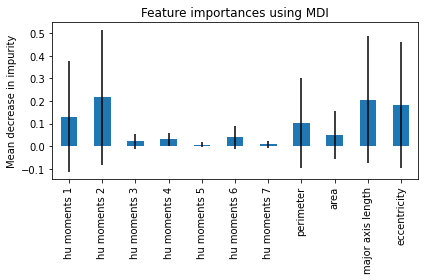
print(forest_importances)
hu moments 1 0.131664
hu moments 2 0.215949
hu moments 3 0.021450
hu moments 4 0.031299
hu moments 5 0.006485
hu moments 6 0.039046
hu moments 7 0.009430
perimeter 0.102604
area 0.051952
major axis length 0.206005
eccentricity 0.184116
dtype: float64
Hu momentsの2つ目とeccentricityが重要だということがわかったので、この2つをx軸、y軸にとり学習用データセットをプロットしてみます。
赤はつまり3粒莢、青は2粒莢を表しています
plt.figure(figsize=(6, 4))
plt.scatter(new_X_train[np.where(y_train == 1.0), 1], new_X_train[np.where(y_train == 1.0), 9], c="red")
plt.scatter(new_X_train[np.where(y_train == 0.0), 1], new_X_train[np.where(y_train == 0.0), 9], c="blue")
plt.xlabel(feature_names[1])
plt.ylabel(feature_names[9])
plt.show()
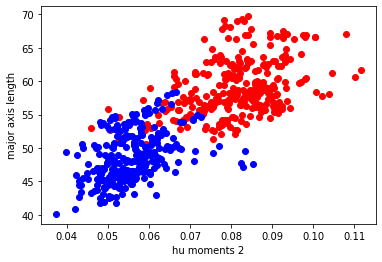
確かに、2つの特徴量だけでも結構うまく分離できそうなことがわかります。
まとめ
今回は「野菜を自動仕分けするAIマシン製作奮闘記 IT農家のラズパイ製ディープ・ラーニング・カメラ」にて紹介されている枝豆2粒莢・3粒莢分類タスクをランダムフォレストフォレストで分類することを考えてみました。
ランダムフォレストなど、ディープラーニング以外の機械学習手法を画像分類に適用する場合、最初に画像からいくつかの特徴量を抽出する必要がありました。今回のタスクと入力画像は比較的単純であり背景が統一されていてきれいだったために簡単に特徴量を抽出することができ、結果的にランダムフォレストでの分類ができました。
また、ランダムフォレストなどの手法はディープラーニングに比べ計算コストが低いためCPUでの計算が簡単にできます。
実際の現場では両者の特性を理解し適材適所で活用していくのが重要だと感じました。
参考
CQ文庫シリーズ 野菜を自動仕分けするAIマシン製作奮闘記 IT農家のラズパイ製ディープ・ラーニング・カメラ 小池誠 著 2020 CQ出版株式会社
GitHub - workpiles/soybeans_sorter: 枝豆選別の実験